年医疗器械生产企业记录表格资料
2025-01-16
精品文档 精品文档 质量记录表 版次 /: A/0 依据 YY/T0287-2017&ISO13485:2016 标准 《医疗器械生产质量管理规范》 编 制: 审 核: 批 准: 2017 -10-10 发布 2017-10-20实施 精品文档 精品文档 质量记录目录 序 号 程序文件 记 录 名 称 文件编号 责任 部门 1 文件控制程序 部门受控文件清单 综合部 文件发放、回收记录 文件借阅、复制记录 文件更改申请 文件销毁申请 文件替换、撤销申请单 外来文件清单 文件更改、销毁、留用记录表 文件归档登记表 2 记录控制程序 文件发放、回收记录 综合部 文件借阅、复制记录 文件销毁申请表 文件归档登记表 3 管理评审控制程 序 管理评审计划 质保部 管理评审会议签到表 管理评审通知单 管理评审报告 管理评审会议记录 质量管理体系改进计划表 4 人力资源控制程 序 年度员工培训计划


采购记录表 年no 产品名称规格型号注册证号单位数量单价金额生产厂家供货单位购货日期采购员备注 验收记录表 年no: 产品名称规格型号注册证号单位数量单价金额生产厂家供货单位购货日期验收员复核员备注 销售记录表 年no: 产品 名称 规格 型号 注册证号单 位

资料 . 受控文件清单(1.质量文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布引 入日期 受控 状态 资料 . 记录: 受控文件清单(2.管理文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布 引入 日期 受控 状态 资料 . 记录: 受控文件清单(3.技术文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布 引入 日期 受控 状态 资料 . 记录: 受控文件清单(4.外来文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 发放号 保管 部门 使用 部门 发布 引入 受控 状态 资料 . 版本号日期 记录:
编辑推荐下载
格式:pdf
大小:1.5MB
页数:101P
人气:92
 4.7
4.7
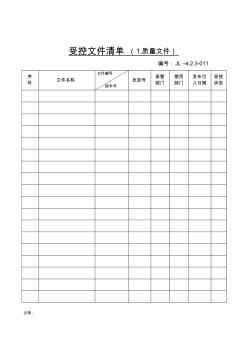
受控文件清单(1.质量文件) 编号:jl–4.2.3-011 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布引 入日期 受控 状态 记录: 受控文件清单(2.管理文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布 引入 日期 受控 状态 记录: 受控文件清单(3.技术文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布 引入 日期 受控 状态 记录: 受控文件清单(4.外来文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布 引入 日期 受控 状态 记录: 文件发放回收记录表编号:jl-4.2.3-02 序号文件名称编号 发 放 号 发放记录回收记录 部门
格式:pdf
大小:1.2MB
页数:14P
人气:92
4.5

产品出库、复核、销售记录 销售规格生产批灭菌批有效 数量生产厂家 质量 购货单位产品名称 号号期至 复核员 日期型号状况 1 产品购进记录 购进 产品名称 规格生产批灭菌批 生产单位经办人供货单位数量 号 效期 日期型号号 2 产品销售记录 销售 产品名称 规格生产批灭菌批 生产单位经办人客户名称数量 号 效期 日期型号号 3 产品验收记录 到货 产品名称 规格生产注册灭菌 效期 质量验收验收 供货单位 型号 数量 证号批号 生产单位 结论人日期批号状况 4 出库单 购货单位:日期: 购货单位产品名称规格型号数量生产日期灭菌日期生产厂家注册证号质量情况有效期 保管员:复核员: 入库单 制单日期: 产品名称规格型号数量生产厂家生产批号灭菌批号注册证号有效期 验收员签字: 5 商品投诉、质量查询报
热门文档 年医疗器械生产企业记录表格资料
格式:pdf
大小:239KB
页数:16P
人气:92
4.5
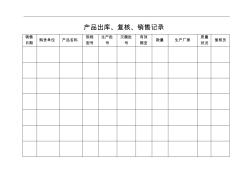
产品出库、复核、销售记录 销售 日期 购货单位产品名称 规格 型号 生产批 号 灭菌批 号 有效 期至 数量生产厂家 质量 状况 复核员 产品购进记录 购进 日期 供货单位产品名称 规格 型号 数量 生产批 号 灭菌批 号 效期生产单位经办人 产品销售记录 销售 日期 客户名称产品名称 规格 型号 数量 生产批 号 灭菌批 号 效期生产单位经办人 产品验收记录 到货 日期 供货单位产品名称 规格 型号 数量 生产 批号 注册 证号 灭菌 批号 效期生产单位 质量 状况 验收 结论 验收 人 出库单 购货单位:日期: 购货单位产品名称规格型号数量生产日期灭菌日期生产厂家注册证号质量情况有效期 保管员:复核员: 入库单 制单日期: 产品名称规格型号数量生产厂家生产批号灭菌批号注册证号有效期 验收员签字: 商品投诉、质量查询报告单 日期客

格式:pdf
大小:6KB
页数:2P
人气:92
4.5

第三类医疗器械生产企业许可证 核发第三类医疗器械生产企业许可证 审批依据:《医疗器械监督管理条例》(国务院令第276号)、《医疗器械生产监督管理办法》 (国家食品药品监督管理局第12号令) 申报条件: 1、企业的生产、质量和技术负责人应当具有与所生产医疗器械相适应的专业能力,并掌握 国家有关医疗器械监督管理的法律、法规和规章以及相关产品质量、技术的规定。质量负责 人不得同时兼任生产负责人; 2、企业内初级以上职称或者中专以上学历的技术人员占职工总比例应当与所生产产品的要 求相适应; 3、企业应当具有与所生产产品及生产规模相适应的生产设备,生产。仓储场地和环境。企 业生产对环境和设备等有特殊要求的医疗器械的,应当符合国家标准、行业标准和国家有关 规定; 4、企业应当设立质量检验机构,并具备与所生产品种和生产规模相适应的质量检验能力; 5、企业应当保存与医疗器械生产和经营有关
格式:pdf
大小:2.0MB
页数:145P
人气:92
4.8

受控文件清单(1.质量文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布引 入日期 受控 状态 记录: 受控文件清单(2.管理文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布 引入 日期 受控 状态 记录: 受控文件清单(3.技术文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布 引入 日期 受控 状态 记录: 受控文件清单(4.外来文件) 编号:jl–4.2.3-01 序 号 文件名称 文件编号 发放号 保管 部门 使用 部门 发布 引入 受控 状态 版本号日期 记录: 文件发放回收记录表编号:jl-4.2.3-02 序号
格式:pdf
大小:41KB
页数:3P
人气:92
4.3

巡查时间:年月日 巡查 项目正常异常原因及处理情况 设备(干燥机附属设施及雨棚、4台 空调散热器、) 完好 门窗、夹层百叶窗完好 消防设施完好 玻璃大厅完好 照明、灭蝇灯、压差表、传递窗完好 设备、桌椅洁净、完好、摆放有序 门窗 洁净、完好(包括玻璃及 锁子) 地面洁净、无新增划痕 空调控制开关(3台)完好 臭氧发生器(3台)洁净、完好 线槽完好 电箱 洁净、完好、停用时处于 关闭状态 设备(纯水机、空压机)、照明 完好、不用时处于关闭状 态 桌椅、检查仪器、玻璃容器、洁具洁净、摆放整齐 给排水阀门及管道完好 门窗完好 电箱、空开、传递窗洁净、完好、不用时关闭 清洗池洁净、完好 给排水阀门及管道洁净、完好 容器、桌椅洁净、摆放整体有序 发生器 洁净、完好、不用时拔掉 插头 雨具架、更鞋凳完好、洁净、摆放整体 饮水机 洁净、完
精华文档 年医疗器械生产企业记录表格资料
格式:pdf
大小:82KB
页数:5P
人气:92
4.4

三类医疗器械分类目录 6804眼科手术器械 -6 眼科手术用其他器 械 玻璃体切割器ⅲ 6815注射穿刺器械 注射穿刺器械 一次性使用无菌注射器及其胶塞、一次性使用无菌注射针、 一次性静脉输液针、一次性使用光纤针、静脉留置针、一 次性配药用注射针、穿刺针 ⅲ 6821医用电子仪器设备 1用于心脏的治疗、急救装置 植入式心脏起搏器、体外心脏起搏器、心脏除颤器、心 脏调搏器、主动脉内囊反搏器、心脏除颤起搏仪 ⅲ 2 有创式电生理仪器及创新电 生理仪器 体外震波碎石机、病人有创监护系统、颅内压监护仪、 有创心输出量计、有创多导生理记录仪、心内希氏束电 图机、心内外膜标测图仪、有创性电子血压计 ⅲ 3有创医用传感器各种植入体内的医用传感器ⅲ 9无创监护仪器心率失常分析仪及报警器、带s-t段的监护仪ⅲ 11医用刺激器心脏工作站电刺激器ⅲ 16体外反搏及其辅助循
格式:pdf
大小:415KB
页数:25P
人气:92
4.5

采购记录表 年no 产品名称规格型号注册证号单位数量单价金额生产厂家供货单位购货日期采购员备注 验收记录表 年no: 产品名称规格型号注册证号单位数量单价金额生产厂家供货单位购货日期验收员复核员备注 销售记录表 年no: 产品 名称 规格 型号 注册证号单 位
格式:pdf
大小:415KB
页数:26P
人气:92
4.5

采购记录表 年no 验收记录表 年no: 产品名称规格型号注册证号单位数量单价金额生产厂家供货单 销售记录表 年no: 产品名称规格型号注册证号单位数量单价金额生产厂家供货单 出库记录表 年
格式:pdf
大小:14KB
页数:1P
人气:92
4.7

医疗器械日常监管现场检查记录 受检单位 检查地址 法定代表人负责人 联系电话手机 监督检查部门 监督检查类别□现场验收□专项检查□飞行检查□案件查处 检查时间年月日 监督检查记录: 1、 检查结论:符合□基本符合√责令改正□限期整改□移交行政处罚□ 检查人员签名: 2015年12月24日 检查情况事实确认意见: 受检企业法定代表人或负责人签名盖章 年月日
格式:pdf
大小:7KB
页数:2P
人气:92
4.6

医疗器械生产企业许可证申请材料要求 《医疗器械生产企业许可证》申请材料要求一、根据核发《医疗器械生产企业许可证》和 变更《医疗器械生产企业许可证》有关事项中的要求提交相关申请材料二、总体要求:(一) 各类表格的内容按填表说明的要求认真填写。(二)申报材料应完整、规范、真实、清晰, 按目录顺序装订成册,并需制软盘或光盘(企业标识不要贴在盘上)。(三)申报材料中除第 三方材料原件外,均需以a4纸打印。(四)各类申请表应按规定要求的份数提供。(五) 未指明必须提供原件的材料或证件,均可提交复印件,复印件上应加盖企业印章(如有)。(六) 申报材料应同时交所在地设区的市食品药品监督管理局一份(如有补正材料,应在材料审查 合格后补交)。三、材料要求(一)《医疗器械生产企业许可证》(开办)申请表(原件)1、 拟开办企业的名称:应与“名称预先核准通知书”或
最新文档 年医疗器械生产企业记录表格资料
格式:pdf
大小:22KB
页数:2P
人气:92
4.8

核发第二类、第三类医疗器械生产企业许可证 项目名称:核发第二类、第三类医疗器械生产企业许可证 ①核发第二类、第三类医疗器械生产企业许可证 审批依据:《医疗器械监督管理条例》(国务院令第276号)、《医疗器械生产监督管理办 法》(国家食品药品监督管理局第12号令) 申报条件: 1、企业的生产、质量和技术负责人应当具有与所生产医疗器械相适应的专业能力,并 掌握国家有关医疗器械监督管理的法律、法规和规章以及相关产品质量、技术的规定。质量 负责人不得同时兼任生产负责人; 2、企业内初级以上职称或者中专以上学历的技术人员占职工总数的比例应当与所生产 产品的要求相适应; 3、企业应当具有与所生产产品及生产规模相适应的生产设备,生产、仓储场地和环境。 企业生产对环境和设备等有特殊要求的医疗器械的,应当符合国家标准、行业标准和国家有 关规定; 4、企业应当设立质量检验机构,并具备与所
格式:pdf
大小:20KB
页数:2P
人气:92
4.8

第二类、第三类医疗器械生产企业需要满足什么条件 (一)企业的生产、质量和技术负责人应当具有与所生产医疗器 械相适应的专业能力,并掌握(奥咨达医疗器械咨询) 国家有关医疗器械监督管理的法律、法规和规章以及相关产品质 量、技术的规定。质量负责人不得 同时兼任生产负责人; (二)企业内初级以上职称或者中专以上学历的技术人员占职工 总数的比例应当与所生产产品 的要求相适应;(只专注于医疗器械领域) (三)企业应当具有与所生产产品及生产规模相适应的生产设 备,生产、仓储场地和环境。企 业生产对环境和设备等有特殊要求的医疗器械的,应当符合国家 标准、行业标准和国家有关规定; (四)企业应当设立质量检验机构,并具备与所生产品种和生产 规模相适应的质量检验能力; (五)企业应当保存与医疗器械生产和经营有关的法律、法规、 规章和有关技术标准。 开办第三类医疗器械生产企业,除应当符合以上要求外,还应当
格式:pdf
大小:34KB
页数:4P
人气:92
4.7
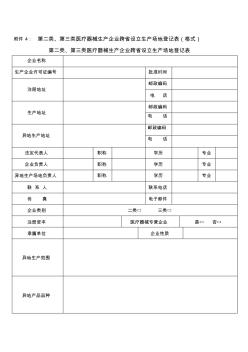
附件4:第二类、第三类医疗器械生产企业跨省设立生产场地登记表(格式) 第二类、第三类医疗器械生产企业跨省设立生产场地登记表 企业名称 生产企业许可证编号批准时间 注册地址 邮政编码 电话 生产地址 邮政编码 电话 异地生产地址 邮政编码 电话 法定代表人职称学历专业 企业负责人职称学历专业 异地生产场地负责人职称学历专业 联系人联系电话 传真电子邮件 企业类别二类□三类□ 注册资本医疗器械专营企业是□否□ 隶属单位企业性质 异地生产范围 异地产品品种 异地生产场地基本情况 职工总数技术人员数 异地生产场所 状况(m2) 建设总面积其中 生产面积净化面积检验面积仓储面积 检验机构状况总人数技术人员数 企业意见 法定代表人签字: 年月日 企业盖章: 年月日 审核意见 签字:年月日 注册地省级(食 品)药品监督管
格式:pdf
大小:1.7MB
页数:116P
人气:92
4.4

受控文件清单(1.质量文件) 编号:jl– 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布引 入日期 受控 状态 记录: 受控文件清单(2.管理文件) 编号:jl– 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布 引入 日期 受控 状态 记录: 受控文件清单(3.技术文件) 编号:jl– 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布 引入 日期 受控 状态 记录: 受控文件清单(4.外来文件) 编号:jl– 序 号 文件名称 文件编号 版本号 发放号 保管 部门 使用 部门 发布 引入 日期 受控 状态 记录: 文件发放回收记录表编号: 序号文件名称编号 发 放 号 发放记录回收记录 部门数量签收人
格式:pdf
大小:26KB
页数:2P
人气:92
4.8

江苏省医疗器械生产企业质量管理体系 (22号令)考核申请表 生产企业:(盖章) 申请目的:□第二、三类产品首次注册 □生产许可证开办 □生产许可证延续 □生产许可证变更-增加产品 □生产许可证变更-生产地址实质性改变 检查标准:□《医疗器械生产企业质量体系考核办法》 江苏省食品药品监督管理局制 江苏省医疗器械生产企业质量管理体系(22号令)考核申请表 一、企业基本情况 企业名称 生产地址1 生产地址2 生产地址3 邮证编码电话传真 法人代表职务职称 管理者代表职务职称 企业管理人员一览表 姓名性别年龄文化程度职务职称主管工作 主要 产品 建厂 日期 占地 面积 平方米建筑面积平方米 职工 总数 人中级职称以上人数人 注册 资金 万元固定资产原值万元 上年
格式:pdf
大小:11KB
页数:3P
人气:92
4.4

医疗器械行业咨询合同 ******有限公司(以下简称“委托方”)计划开展医疗器械产品的研发和制造,特 委托**********(以下简称“受托方”),就“医疗器械企业开办和产品注册等”提供咨 询服务,双方就此签署如下合同: 1委托内容 (1)对委托方公司现有情况进行调研; (2)对委托方公司未来规划进行调研; (3)对委托方公司拟开发的产品进行调研和咨询; (4)对委托方公司医疗器械企业开办提供咨询; (5)对委托方公司医疗器械产品注册或备案提供咨询; (6)对委托方公司医疗器械产品开发流程提供咨询; (7)对委托方公司医疗器械行业法律法规提供培训和咨询; (8)委托方与受托方商议决定的其他咨询内容。 2咨询办法和提供的文件 受托方根据上述项目规定的任务进行调研、培训和咨询;主要通过以下方 式提供服务: 1)通过当面的技术交流和指导,使委托方的公司未来规

格式:pdf
大小:427KB
页数:27P
人气:92
4.7
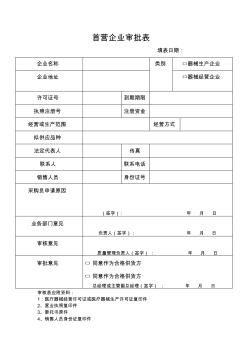
首营企业审批表 填表日期: 企业名称类别□器械生产企业 企业地址□器械经营企业 许可证号到期期限 执照注册号注册资金 经营或生产范围经营方式 拟供应品种 法定代表人传真 联系人联系电话 销售人员身份证号 采购员申请原因 (签字):年月日 业务部门意见 负责人(签字):年月日 审核意见 质量管理负责人(签字):年月日 审批意见□同意作为合格供货方 □同意作为合格供货方 总经理或主管副总经理(签字):年月日 审核表应附资料: 1:医疗器械经营许可证或医疗器械生产许可证复印件 2、营业执照复印件 3、委托书原件 4、销售人员身份证复印件 首营品种审批表 编号:1 供货单位(经营企业)名称及 资质证明、联系方式 医疗器械产品 名称 规格 生产企业名称及 资质证明 许可证号:许可证号: 电话: 医疗器械性能、用途、外观、质量情况审核 注册
格式:pdf
大小:523KB
页数:54P
人气:92
4.6

***********************清洁生产审核报告 1 医疗器械行业清洁生产审核报告 ***********************清洁生产审核报告 1 目录 前言.........................................................................................错误!未定义书签。 1筹划和组织.............................................................................................................1 1.1清洁生产审核工作小组.....................................................................
格式:pdf
大小:480KB
页数:27P
人气:92
4.4
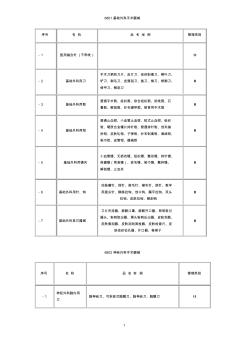
1 6801基础外科手术器械 6803神经外科手术器械 序号名称品名举例管理类别 -1 神经外科脑内用 刀 脑神经刀、可拆卸式脑膜刀、脑神经刀、脑膜刀ⅱ 序号名称品名举例管理类别 -1医用缝合针(不带线)ⅱ -2基础外科用刀 手术刀柄和刀片、皮片刀、疣体剥离刀、柳叶刀、 铲刀、剃毛刀、皮屑刮刀、挑刀、锋刀、修脚刀、 修甲刀、解剖刀 ⅰ -3基础外科用剪 普通手术剪、组织剪、综合组织剪、拆线剪、石 膏剪、解剖剪、纱布绷带剪、教育用手术剪 ⅰ -4基础外科用钳 普通止血钳、小血管止血钳、蚊式止血钳、组织 钳、硬质合金镶片持针钳、普通持针钳、创夹缝 拆钳、皮肤轧钳、子弹钳、纱布剥离钳、海绵钳、 帕巾钳、皮管钳、器械钳 ⅰ -5基础外科用镊夹 小血管镊、无损伤镊、组织镊、整形镊、持针镊、 保健镊(简易镊)、拔毛镊、帕巾镊、敷料镊、 解剖镊、止血
文辑创建者
我要分享 >
职位:信息化咨询工程师
擅长专业:土建 安装 装饰 市政 园林
相关编辑
文辑推荐
知识推荐
百科推荐