综述
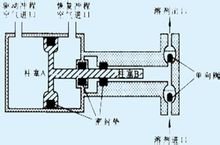
HPLC的出现不过三十多年的时间,但这种分离分析技术的发展十分迅猛,目前应用也十分广泛。其仪器结构和流程也多种多样。典型的高效液相色谱仪结构和流程可用下列方框图表示(See Fig.3-4)。高效液相色谱仪一般都具备贮液器、高压泵、梯度洗提装置(用双泵)、进样器、色谱柱、检测器、恒温器、记录仪等主要部件。
高效液相色谱更适宜于分离、分析高沸点、热稳定性差、有生理活性及相对分子量比较大的物质,因而广泛应用于核酸、肽类、内酯、稠环芳烃、高聚物、药物、人体代谢产物、表面活性剂,抗氧化剂、杀虫剂、除莠剂的分析等物质的分析。
高压泵
HPLC使用的色谱柱是很细的(1~6 mm),所用固定相的粒度也非常小(几μm到几十μm),所以流动相在柱中流动受到的阻力很大,在常压下,流动相流速十分缓慢,柱效低且费时。为了达到快速、高效分离,必须给流动相施加很大的压力,以加快其在柱中的流动速度。为此,须用高压泵进行高压输液。高压、高速是高效液相色谱的特点之一。 HPLC使用的高压泵应满足下列条件:
a. 流量恒定,无脉动,并有较大的调节范围(一般为1~10 mL/min);
b. 能抗溶剂腐蚀;
c. 有较高的输液压力;对一般分离,60×10^5Pa的压力就满足了,对高效分离,要求达到150~300×10^5Pa。
⑴. 往复式柱塞泵
当柱塞推入缸体时,泵头出口(上部)的单向阀打开,同时,流动相进入的单向阀(下部)关闭,这时就输出少量的流体。反之,当柱塞向外拉时,流动相入口的单向阀打开,出口的单向阀同时关闭,一定量的流动相就由其储液器吸入缸体中。这种泵的特点是不受整个色谱体系中其余部分阻力稍有变化的影响,连续供给恒定体积的流动相。
⑵气动放大泵
其工作原理是:压力为 p1 的低压气体推动大面积( SA )活塞A ,则在小面积( SB )活塞 B 输出压力增大至 p2 的液体。压力增大的倍数取决于 A 和 B 两活塞的面积比,如果 A 与 B 的面积之比为 50 : 1 ,则压力为 5 × Pa 的气体就可得到压力为 250×Pa 的输出液体。这是一种恒压泵。
梯度洗提
类似于GC中的程序升温。已成为现代高效液相色谱中不可缺少的部分。 梯度洗提,就是载液中含有两种(或更多)不同极性的溶剂,在分离过程中按一定的程序连续改变载液中溶剂的配比和极性,通过载液中极性的变化来改变被分离组分的分离因素,以提高分离效果。梯度洗提可以分为两种:
a. 低压梯度(也叫外梯度):在常压下,预先按一定程序将两种或多种不同极性的溶剂混合后,再用一台高压泵输入色谱柱。
b.高压梯度 ( 或称内梯度系统 ) :利用两台高压输液泵,将两种不同极性的溶剂按设定的比例送入梯度混合室,混合后,进入色谱柱。
进样装置
⑴.注射器进样装置:进样所用微量注射器及进样方式与 GC法一样。进样压力150×10^5Pa时,必须采用停流进样。⑵.高压定量进样阀:与GC法用的流通法相似,能在高压下进样。
色谱柱
色谱柱是色谱仪最重要的部件(心脏)。通常用厚壁玻璃管或内壁抛光的不锈钢管制作的,对于一些有腐蚀性的样品且要求耐高压时,可用铜管、铝管或聚四氟乙烯管。柱子内径一般为1~6 mm。常用的标准柱型是内径为 4.6 或 3.9mm ,长度为 15 ~ 30cm 的直形不锈钢柱。填料颗粒度 5 ~ 10μm ,柱效以理论塔板数计大约 7000 ~ 10000。
发展趋势是减小填料粒度和柱径以提高柱效。
检测器
⑴.紫外光度检测器
它的作用原理是基于被分析试样组分对特定波长紫外光的选择性吸收,组分浓度与吸光度的关系遵守比尔定律。最常用的检测器,应用最广,对大部分有机化合物有响应。
特点:
a.灵敏度高:其最小检测量10-9g·mL-1,故即使对紫外光吸收很弱的物质,也可以检测;
b. 线性范围宽;(比尔定律)
c. 流通池可做的很小(1mm × 10mm ,容积 8μL);
d. 对流动相的流速和温度变化不敏感可用于梯度洗脱;
e. 波长可选,易于操作:如,使用装有流通池的可见紫外分光光度计(可变波长检测器)。
缺点:对紫外光完全不吸收的试样不能检测;同时溶剂的选择受到限制。
⑵. 光电二极管阵列检测器
紫外检测器的重要进展;阵列由1024个光电二极管阵列,每个光电二极管宽仅50μm,各检测一窄段波长。如图所示,在检测器中,光源发出的紫外或可见光通过液相色谱流通池,在此流动相中的各个组分进行特征吸收,然后通过狭缝,进入单色其进行分光,最后由光电二极管阵列检测,得到各个组分的吸收信号。经计算机快速处理,得三维立体谱图。
⑶.荧光检测器
荧光检测器是一种高灵敏度、高选择性检测器。
对多环芳烃,维生素B、黄曲霉素、卟啉类化合物、农药、药物、氨基酸、甾类化合物等有响应。
荧光检测器的结构及工作原理和荧光光度计相似。
⑷.差示折光检测器
除紫外检测器之外应用最多的检测器。
差示折光检测器是借连续测定流通池中溶液折射率的方法来测定试样浓度的检测器。溶液的折射率是纯溶剂(流动相)和纯溶质(试样)折射率乘以各物质的浓度之和。因此溶有试样的流动相和纯流动相之间折射率之差表示试样在流动相中的浓度。
⑸.电导检测器
其作用原理是根据物质在某些介质中电离后所产生电导变化来测定电离物质含量。
-