自硬呋喃树脂呋喃树脂的固化文献

 呋喃树脂生产工艺规程
呋喃树脂生产工艺规程
呋喃树脂生产工艺规程
呋喃树脂生产工艺规程 1、产品说明 名称:呋喃树脂 ; 分子式: C5H7-〔C7H11N2O〕n-C5H7O 结构式: 分子量: 289-845 物理性质:黄棕色透明液体,有轻微的刺激性气味, 无毒。粘度(200C) 15-30CP,比重( 200C)1.19g/ml , pH值,对皮肤 有轻 微刺激作用。 化学性质:与酸类物质能发生剧烈的放热反应, 生成不溶性的沥青状 固体物。 质量标准: JB/T 7527-94 包装与储运要求:铁桶包装,净重 240Kg/桶。密闭储存于阴凉干燥 处,不能暴晒,远离热源,严禁接触酸性物质,按一般化学品规 定运输。 主要用途:在铸造行业用作自硬树脂砂的粘结剂。 2、原料质量标准及包装要求 甲醛: 2.1.1质量标准 GB9009-88 2.1.2 理化性质:无色透明液体,有刺激性气味,相对密度,反应活 性强,易聚合。工业品中一般加 8-12%的甲醇作阻
 铸造用呋喃树脂砂
铸造用呋喃树脂砂
铸造用呋喃树脂砂
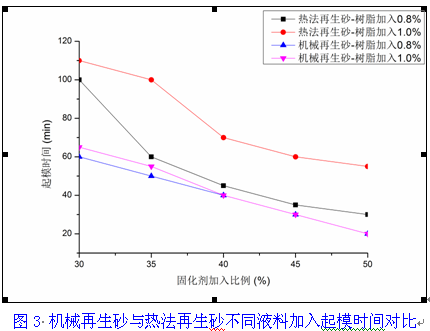
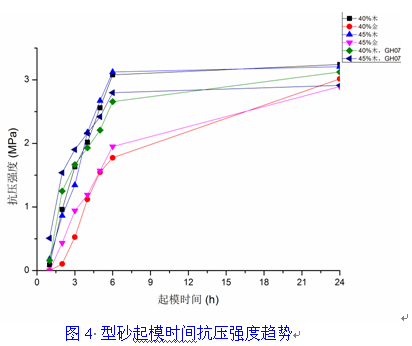
第一章 铸造用呋喃树脂砂概述 一、自硬呋喃树脂砂的特点 1. 优点 : 1) 铸件表面光洁、棱角清晰、尺寸精度高; 2) 型砂的溃散性好,清理、打磨容易,从而减少了落砂清铲修整工序中对铸 件形状精度的损害; 3) 由于在各个工序中都最大限度的排除了影响铸型、铸件变形和损坏的因 素,所以树脂砂铸件的铸件表面质量、 铸件几何尺寸精度方面比黏土可以 提高 1~2 级,达到了 CT7~9 级精度和 1~2mm/600mm 的平直度,表面粗 糙度大有改观; 4) 减轻劳动强度 大大改善了劳动条件和工作环境, 尤其是减轻了噪声、 矽 尘等,减少了环境污染; 5) 树脂砂型 (芯)强度高 (含高温强度高 )、成型性好 发气量较其它有机铸型 低、热稳定性好、透气性好,可以大大减少铸件的粘砂、夹砂、砂眼、气 孔、缩孔、裂纹等铸件缺陷,从而降低废品率,可以制造出用黏土砂难以 做出的复杂件、关键件; 6) 旧